Blinda's Class
Classwork for BIMM143
Lab 11 Structural Bioinformatics pt2
Blinda Sui (PID: A17117043)
AlphaFold Data Base (AFDB)
The EBI maintains the larest database of AlphaFold structure prediction models at: http://alphafold.ebi.ac.uk
From last class (before Halloween) we saw that the PDB had 244,290 (Oct 2025)
The total number of protein sequences in UniProtKB is 199,579,901
Key Point: This is a tiny fraction of sequence space that has structural coverage (0.12%)
244290/199579901 * 100
[1] 0.1224021
AFDB is attempting to address this gap…
There are two “Quality Scores” from AlphaFold one for residues (i.e. each amino acid) called pLDDT score. The other PAE score that measures the confidence in the relative position of two residues (i.e. a score for every pair of residues).
Generating your own structure predictions
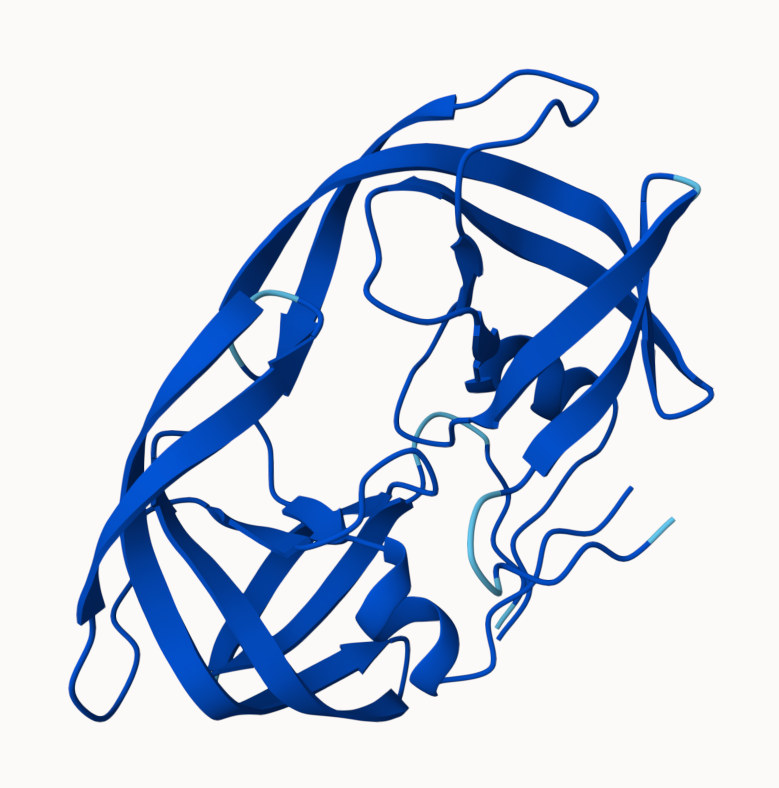
Figure of 5 generated HIP-PR models
![]()
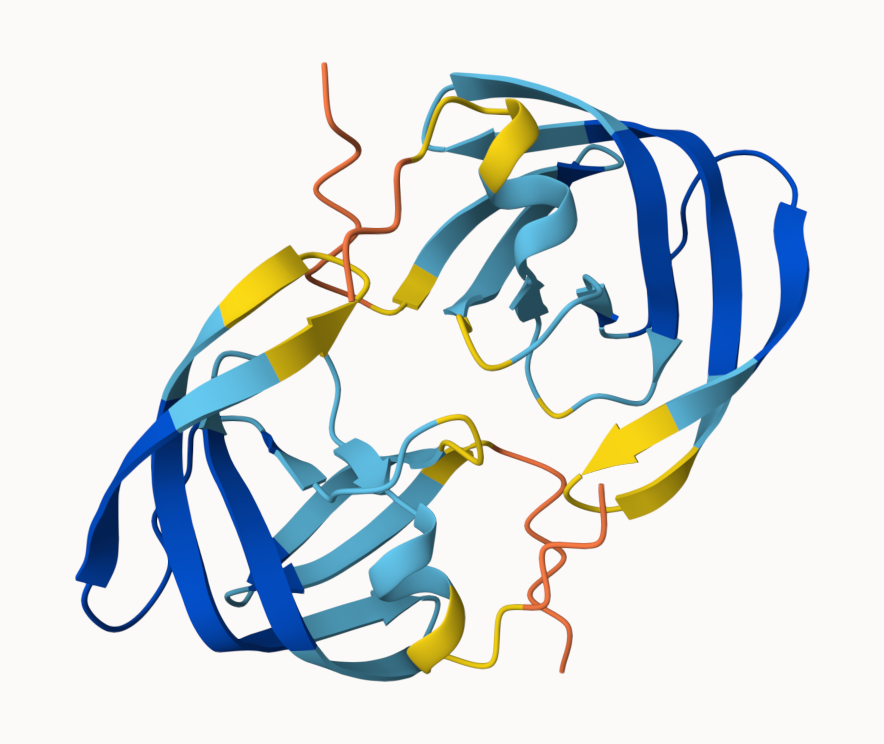
and the top ranked model colored by chain
![]()
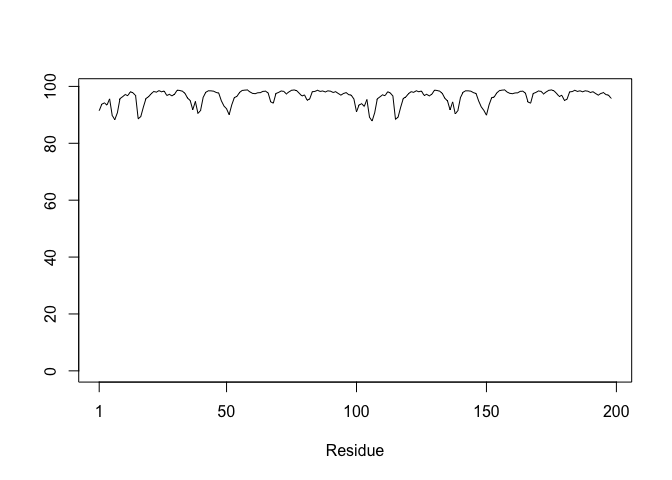
pLDDT score for model 1
and model 5
Custom analysis of resulting models in R
Read key result files into R. The first thing I need to know is what my results direcvtory/folder is called (i.e. it name is different for every AlphaFold run/job)
results_dir <- "HIPPR_dimer_23119/"
# File names for all PDB models
pdb_files <- list.files(path=results_dir,
pattern="*.pdb",
full.names = TRUE)
# Print our PDB file names
basename(pdb_files)
[1] "HIPPR_dimer_23119_unrelaxed_rank_001_alphafold2_multimer_v3_model_4_seed_000.pdb"
[2] "HIPPR_dimer_23119_unrelaxed_rank_002_alphafold2_multimer_v3_model_1_seed_000.pdb"
[3] "HIPPR_dimer_23119_unrelaxed_rank_003_alphafold2_multimer_v3_model_5_seed_000.pdb"
[4] "HIPPR_dimer_23119_unrelaxed_rank_004_alphafold2_multimer_v3_model_2_seed_000.pdb"
[5] "HIPPR_dimer_23119_unrelaxed_rank_005_alphafold2_multimer_v3_model_3_seed_000.pdb"
library(bio3d)
m1 <- read.pdb(pdb_files[1])
m1
Call: read.pdb(file = pdb_files[1])
Total Models#: 1
Total Atoms#: 1514, XYZs#: 4542 Chains#: 2 (values: A B)
Protein Atoms#: 1514 (residues/Calpha atoms#: 198)
Nucleic acid Atoms#: 0 (residues/phosphate atoms#: 0)
Non-protein/nucleic Atoms#: 0 (residues: 0)
Non-protein/nucleic resid values: [ none ]
Protein sequence:
PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYD
QILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNFPQITLWQRPLVTIKIGGQLKE
ALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYDQILIEICGHKAIGTVLVGPTP
VNIIGRNLLTQIGCTLNF
+ attr: atom, xyz, calpha, call
plot(m1$atom$b[m1$calpha], typ="l")
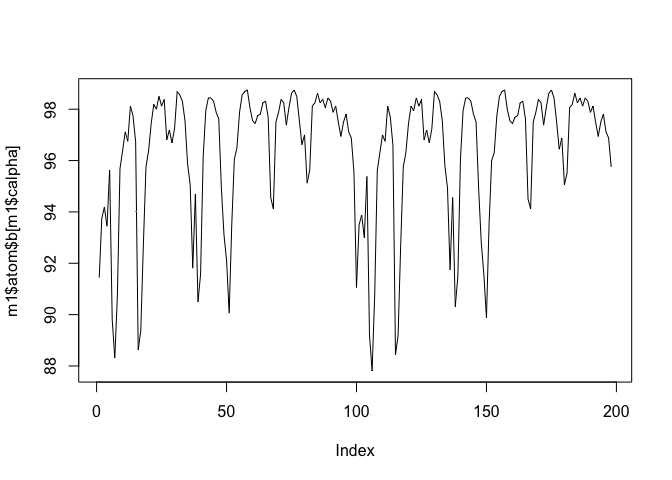
plot.bio3d(m1$atom$b[m1$calpha], typ="l")
Residue conservation from alignment file
Find the large AlphaFold alignment file
aln_file <- list.files(path=results_dir,
pattern=".a3m$",
full.names = TRUE)
aln_file
[1] "HIPPR_dimer_23119//HIPPR_dimer_23119.a3m"
Read this into R
aln <- read.fasta(aln_file[1], to.upper = TRUE)
[1] " ** Duplicated sequence id's: 101 **"
[2] " ** Duplicated sequence id's: 101 **"
How many sequences are in this alignment
dim(aln$ali)
[1] 5397 132
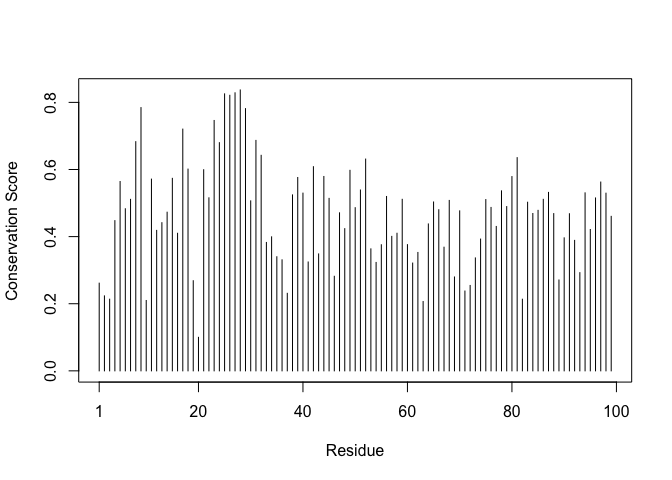
We can score residue conservation in the alignment with the conserv() function.
sim <- conserv(aln)
plotb3(sim[1:99], ylab="Conservation Score")
con <- consensus(aln, cutoff = 0.9)
con$seq
[1] "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-"
[19] "-" "-" "-" "-" "-" "-" "D" "T" "G" "A" "-" "-" "-" "-" "-" "-" "-" "-"
[37] "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-"
[55] "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-"
[73] "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-"
[91] "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-"
[109] "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-"
[127] "-" "-" "-" "-" "-" "-"