Blinda's Class
Classwork for BIMM143
Lab 12 DESeq Lab
Blinda Sui (PID: A17117043)
- Background
- Data import
- Toy differential gene expression
- DESeq2 analysis
- Volcano Plot
- Save our results
Background
Today we will analyze some RNASeq data from Himes et al.on the effects of a common steroid (dexamethasone) on airway smooth muscle cells (ASM cells)
Are starting point is the “counts” data and “metadata” that contain the count values for each gene in their different experiments (i.e. cell lines with or without the drug)
Data import
#Complete the missing code
counts <- read.csv("airway_scaledcounts.csv", row.names=1)
metadata <- read.csv("airway_metadata.csv")
head(counts)
SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516
ENSG00000000003 723 486 904 445 1170
ENSG00000000005 0 0 0 0 0
ENSG00000000419 467 523 616 371 582
ENSG00000000457 347 258 364 237 318
ENSG00000000460 96 81 73 66 118
ENSG00000000938 0 0 1 0 2
SRR1039517 SRR1039520 SRR1039521
ENSG00000000003 1097 806 604
ENSG00000000005 0 0 0
ENSG00000000419 781 417 509
ENSG00000000457 447 330 324
ENSG00000000460 94 102 74
ENSG00000000938 0 0 0
Q1. How many genes are in this dataset?
nrow(counts)
[1] 38694
Q. How many different experiments (columns in counts or rows in metadata) are there?
nrow(metadata)
[1] 8
Q2. How many ‘control’ cell lines do we have?
sum(metadata$dex == "control")
[1] 4
Toy differential gene expression
To start our analysis let’s calculate the mean counts for all genes in the “control” experiments.
- Extract all “control” columns from the
countsobject. - Calculate the mean for all rows (i.e. gnes) of these “control” columns. 3-4. Do the same for “treated”.
- Compare these
control.meanandtreated.meanvalues.
#1
control.inds <- metadata$dex == "control"
control.counts <- counts[,control.inds]
Q3. How would you make the above code in either approach more robust? Is there a function that could help here?
rowMean()
Q4. Follow the same procedure for the treated samples (i.e. calculate the mean per gene across drug treated samples and assign to a labeled vector called treated.mean)
#2
control.means <- rowMeans(control.counts)
#3-4
treated.means <- rowMeans(counts[, metadata$dex == "treated"])
Store these together for case of bookkeeping as meancounts
#5
meancounts <- data.frame(control.means, treated.means)
head(meancounts)
control.means treated.means
ENSG00000000003 900.75 658.00
ENSG00000000005 0.00 0.00
ENSG00000000419 520.50 546.00
ENSG00000000457 339.75 316.50
ENSG00000000460 97.25 78.75
ENSG00000000938 0.75 0.00
Make a plot of control vs treated mean values ofr all genes
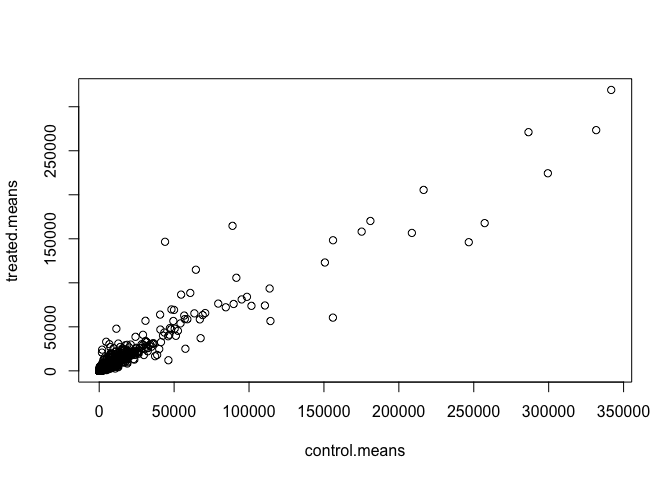
Q5 (a). Create a scatter plot showing the mean of the treated samples against the mean of the control samples. Your plot should look something like the following.
plot(meancounts)
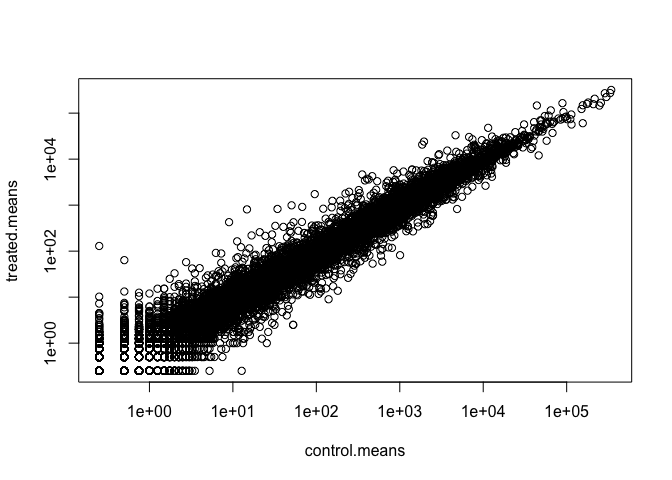
plot(meancounts, log="xy")
Warning in xy.coords(x, y, xlabel, ylabel, log): 15032 x values <= 0 omitted
from logarithmic plot
Warning in xy.coords(x, y, xlabel, ylabel, log): 15281 y values <= 0 omitted
from logarithmic plot
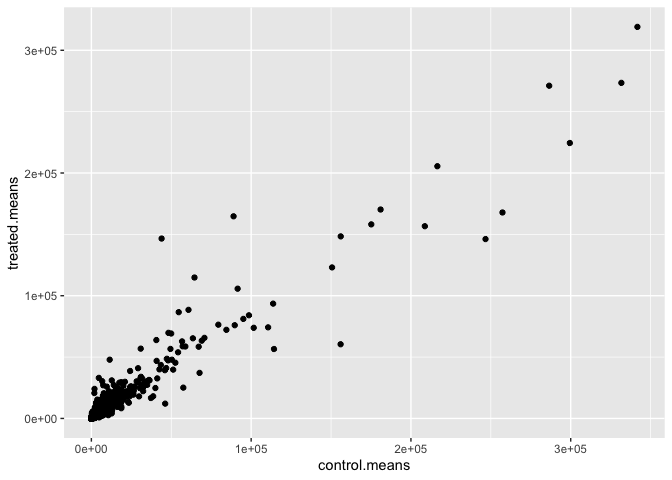
Q5 (b).You could also use the ggplot2 package to make this figure producing the plot below. What geom_?() function would you use for this plot?
library(tidyverse)
── Attaching core tidyverse packages ──────────────────────── tidyverse 2.0.0 ──
✔ dplyr 1.1.4 ✔ readr 2.1.5
✔ forcats 1.0.1 ✔ stringr 1.5.2
✔ ggplot2 4.0.0 ✔ tibble 3.3.0
✔ lubridate 1.9.4 ✔ tidyr 1.3.1
✔ purrr 1.1.0
── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()
ℹ Use the conflicted package (<http://conflicted.r-lib.org/>) to force all conflicts to become errors
ggplot(meancounts, aes(x = control.means, y = treated.means)) +
geom_point()
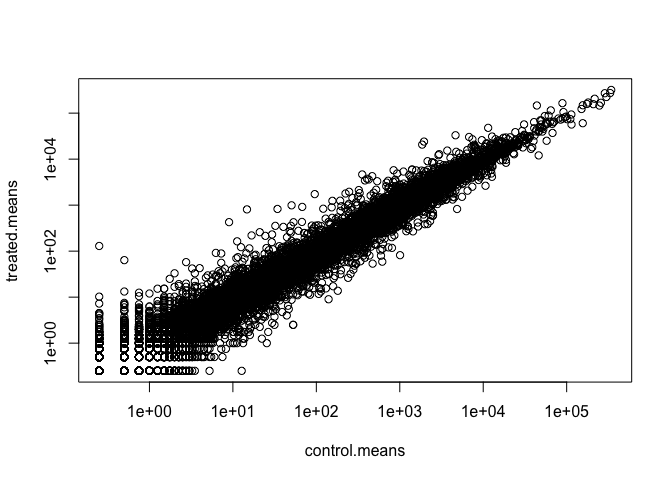
Q6. Try plotting both axes on a log scale. What is the argument to plot() that allows you to do this?
plot(control.means, treated.means, log = "xy")
Warning in xy.coords(x, y, xlabel, ylabel, log): 15032 x values <= 0 omitted
from logarithmic plot
Warning in xy.coords(x, y, xlabel, ylabel, log): 15281 y values <= 0 omitted
from logarithmic plot
Q7. What is the purpose of the arr.ind argument in the which() function call above? Why would we then take the first column of the output and need to call the unique() function?
The arr.ind=TRUE argument will clause which() to return both the row and column indices (i.e. positions) where there are TRUE values. In this case this will tell us which genes (rows) and samples (columns) have zero counts. We are going to ignore any genes that have zero counts in any sample so we just focus on the row answer. Calling unique() will ensure we don’t count any row twice if it has zero entries in both samples.
We often talk metrics like “log2 fold-change”
# treated/control
log2(10/10)
[1] 0
log2(10/20)
[1] -1
log2(20/10)
[1] 1
log2(40/10)
[1] 2
log2(10/40)
[1] -2
Let’s calculate the log2 fold change for our treated over control mean counts.
meancounts$log2fc <- log2(meancounts$treated.means/
meancounts$control.means)
head(meancounts)
control.means treated.means log2fc
ENSG00000000003 900.75 658.00 -0.45303916
ENSG00000000005 0.00 0.00 NaN
ENSG00000000419 520.50 546.00 0.06900279
ENSG00000000457 339.75 316.50 -0.10226805
ENSG00000000460 97.25 78.75 -0.30441833
ENSG00000000938 0.75 0.00 -Inf
A common “rule of thumb” is a log2 fold change cutoff +2 and -2 to call genes “Up regulated” or “Down regulated”.
sum(meancounts$log2fc >= +2, na.rm=T)
[1] 1910
#When na.rm is set to TRUE, the function will remove or ignore any NA values present in the input data before performing the calculation. If na.rm is set to FALSE (which is often the default), and there are NA values in the data, the function will typically return NA as the result, as it cannot compute a meaningful value when missing data is present.
Number of “down” genes at -2 threshold
sum(meancounts$log2fc <= -2, na.rm=T)
[1] 2330
Q8. Using the up.ind vector above can you determine how many up regulated genes we have at the greater than 2 fc level?
Q9. Using the down.ind vector above can you determine how many down regulated genes we have at the greater than 2 fc level?
up.ind <- meancounts$log2fc > 2
down.ind <- meancounts$log2fc < (-2)
sum(up.ind, na.rm=T)
[1] 1846
sum(down.ind, na.rm=T)
[1] 2212
Q10. Do you trust these results? Why or why not?
This fold change is not enough to determin the significance, so I do not trust thesed results.
DESeq2 analysis
Let’s do this analysis properly and keep our inner stats nerd happy - i.e. are the differences we see between drug and no drug significant given the replicate experiments.
library(DESeq2)
For DESeq analysis we need three things
- count values (
contdata) - metadata telling us about the columns in
countData(colData) - design of the experiment (i.e. what do you want to compare)
Our first function form DESeq2 will setup the input required for analysis by storing all these 3 things together.
dds <- DESeqDataSetFromMatrix(countData = counts,
colData = metadata,
design = ~dex)
converting counts to integer mode
Warning in DESeqDataSet(se, design = design, ignoreRank): some variables in
design formula are characters, converting to factors
The main function in DESeq2 that runs the analysis is called DESeq()
dds <- DESeq(dds)
estimating size factors
estimating dispersions
gene-wise dispersion estimates
mean-dispersion relationship
final dispersion estimates
fitting model and testing
res <- results(dds)
head(res)
log2 fold change (MLE): dex treated vs control
Wald test p-value: dex treated vs control
DataFrame with 6 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
ENSG00000000003 747.194195 -0.350703 0.168242 -2.084514 0.0371134
ENSG00000000005 0.000000 NA NA NA NA
ENSG00000000419 520.134160 0.206107 0.101042 2.039828 0.0413675
ENSG00000000457 322.664844 0.024527 0.145134 0.168996 0.8658000
ENSG00000000460 87.682625 -0.147143 0.256995 -0.572550 0.5669497
ENSG00000000938 0.319167 -1.732289 3.493601 -0.495846 0.6200029
padj
<numeric>
ENSG00000000003 0.163017
ENSG00000000005 NA
ENSG00000000419 0.175937
ENSG00000000457 0.961682
ENSG00000000460 0.815805
ENSG00000000938 NA
36000 * 0.05
[1] 1800
Volcano Plot
This is a common summary result figure from these types of experiments and plot the log2 fold-change vs the adjusted p-value
plot(res$log2FoldChange, -log(res$padj))
abline(v=c(-2,2), col="red")
abline(h=-log(0.04), col="red")
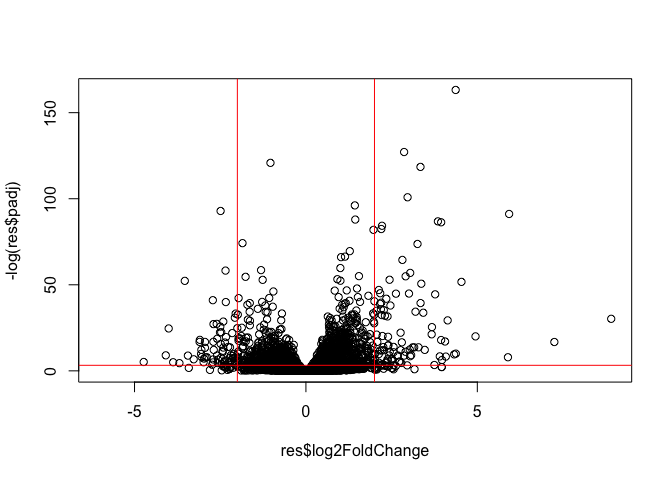
#abline() function in R is used to add one or more straight lines to an existing plot. It is a powerful tool for enhancing data visualization by highlighting trends, relationships, or specific reference points within your data.
Save our results
write.csv(res, file="my_results.csv")